BETA-THALASSEMIA PAGE
Understanding Thalassemia
Thalassemia is a group of inherited blood disorders caused by mutations in the genes that control hemoglobin production. Hemoglobin is the protein in red blood cells responsible for carrying oxygen throughout the body. When hemoglobin is not produced correctly, red blood cells become fragile, leading to chronic anemia and a host of related complications.
There are two main types of thalassemia, classified by which component of the hemoglobin molecule is affected:
- Alpha Thalassemia: Involves mutations in the alpha-globin genes.
- Beta Thalassemia: Involves mutations in the beta-globin genes.
What is Beta-Thalassemia?
Beta-Thalassemia is the most prevalent and clinically significant form of thalassemia worldwide. It results from mutations in the HBB gene, which encodes the beta-globin chains of hemoglobin. This leads to reduced or absent production of functional beta-globin, disrupting the balance of hemoglobin formation and causing ineffective red blood cell production and anemia.
Beta-Thalassemia is inherited in an autosomal recessive pattern. A person who inherits two mutated copies of the HBB gene—one from each parent—will have a more severe form of the disease.
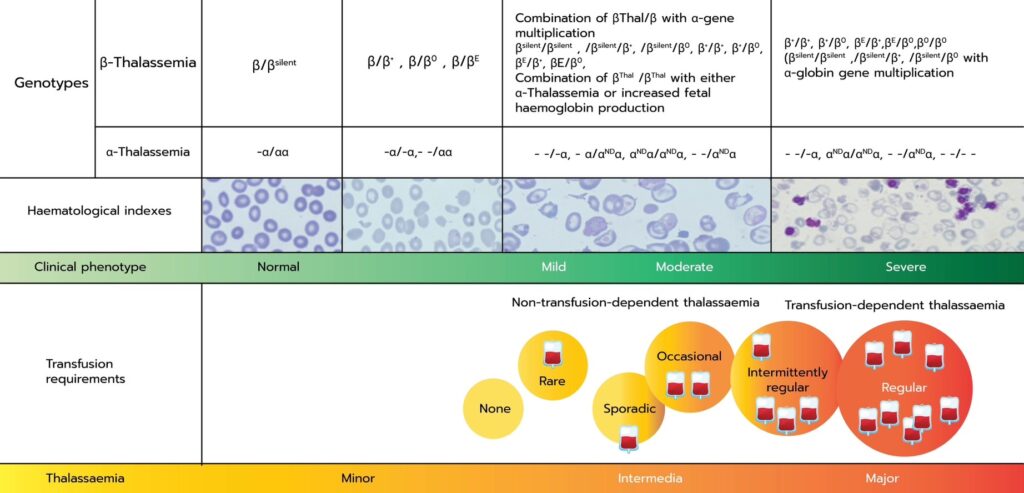
There are three clinical classifications of beta-thalassemia:
- Beta-Thalassemia Minor: A carrier state with no symptoms or mild anemia.
- Beta-Thalassemia Intermedia: Moderate anemia that may not require regular transfusions.
- Beta-Thalassemia Major (Cooley’s Anemia): Severe anemia requiring lifelong blood transfusions—also known as Transfusion-Dependent Thalassemia (TDT).
Transfusion-Dependent Thalassemia (TDT)
Transfusion-Dependent Thalassemia (TDT) is the most severe form of beta-thalassemia. Patients with TDT are unable to produce sufficient hemoglobin to sustain normal red blood cell levels and require regular blood transfusions, often every 2–5 weeks, starting in early childhood.
While transfusions are life-saving, they are not curative. Over time, they lead to iron overload, which can cause severe damage to the heart, liver, and endocrine system. Patients with TDT often require iron chelation therapy to manage iron buildup, as well as ongoing medical surveillance and treatment of complications.
Common Complications of TDT:
- Iron-induced cardiomyopathy and heart failure
- Liver fibrosis and cirrhosis
- Endocrine dysfunction (e.g., diabetes, growth failure, infertility)
- Bone deformities and osteoporosis
- Increased risk of infections and transfusion-related complications
The physical, emotional, and economic burden of lifelong transfusions is profound, impacting quality of life for patients and families. Despite improvements in supportive care, life expectancy remains reduced in many regions, especially where access to high-quality transfusion and chelation therapy is limited.
A Genetic Disease with a Global Impact
TDT is most prevalent in regions with historically high rates of malaria, where thalassemia gene mutations provided some evolutionary protection against infection. As a result, thalassemia is common in parts of:
- Southeast Asia
- South Asia
- the Middle East
- the Mediterranean
- Africa
With increasing global migration, thalassemia is now a worldwide concern. The World Health Organization has identified it as a major public health challenge, particularly in low- and middle-income countries, where access to curative therapies remains limited.
Economic and Social Burden of Transfusion-Dependent Thalassemia
Transfusion-Dependent Thalassemia (TDT) imposes a significant and lifelong burden—not only on individual patients and families, but also on healthcare systems and broader societies. Beyond the clinical challenges, the financial, logistical, and emotional demands of managing this chronic genetic disease are substantial and often underestimated.
Direct Medical Costs
The cornerstone of TDT management is lifelong, regular blood transfusions and iron chelation therapy. These treatments, while essential, are expensive and resource-intensive:
- Blood transfusions require compatible blood supplies, rigorous safety screening, trained personnel, and clinical infrastructure.
- Iron chelation medications, needed to prevent life-threatening organ damage, can cost thousands of dollars per year per patient and require long-term adherence.
- Monitoring and treating complications—including heart disease, liver dysfunction, endocrine failure, and infections—add to the cumulative healthcare cost.
In many countries, TDT treatment consumes a disproportionate share of public healthcare budgets, especially in regions with high disease prevalence and limited healthcare infrastructure.
Indirect Economic Costs
The financial impact of TDT extends well beyond the hospital:
- Loss of productivity due to frequent medical visits, hospital stays, and chronic fatigue affects both patients and caregivers.
- Caregiver burden is substantial, particularly for children with TDT, often requiring parents or family members to reduce work hours or leave employment altogether.
- Education disruption is common, with young patients missing school frequently due to transfusions or complications.
These cumulative indirect costs hinder economic independence, career development, and social mobility, especially in lower-income households and underserved regions.
Emotional and Psychosocial Impact
Living with TDT is not only physically taxing but emotionally demanding. Patients often face:
- Chronic anxiety over long-term complications and survival.
- Social stigma associated with visible symptoms, regular hospital visits, or cultural misconceptions about inherited diseases.
- Mental health challenges, including depression, isolation, and reduced quality of life—especially in adolescence and young adulthood.
The psychosocial toll affects not just individuals, but entire families and communities, reinforcing cycles of emotional and financial hardship.
Health Inequity and Global Disparity
Access to high-quality TDT care is uneven across the globe. In many low- and middle-income countries, patients face:
- Limited availability of safe blood supplies
- High out-of-pocket costs for chelation therapy
- Delays in diagnosis and treatment
- Lack of specialized care or transplant options
These disparities highlight the urgent need for affordable, scalable, and curative solutions that reduce long-term dependency on medical systems and improve health equity.
A Need for Transformative Therapies
Current standard-of-care therapies—including transfusion and chelation—are not curative and require lifelong adherence, creating a heavy burden for patients. Allogeneic bone marrow transplantation is the only established curative option, but it is limited by donor availability, risks of graft-versus-host disease (GVHD), and transplant-related complications.
This unmet medical need has driven the development of gene therapy as a promising curative approach. By correcting or compensating for the faulty beta-globin gene in a patient’s own cells, gene therapy offers the potential to restore normal hemoglobin production and eliminate transfusion dependency.
Nouvogenes Therapeutics’ Mission
At Nouvogenes Therapeutics, we are developing CRISPR-based gene therapies with the potential to cure Transfusion-Dependent Thalassemia by addressing the genetic root cause of the disease. Our goal is to offer a durable, one-time treatment that can free patients from lifelong transfusions, prevent long-term complications, and improve quality of life.
We believe that patients around the world—regardless of geography or socioeconomic background—deserve access to next-generation cures that offer true hope, not just disease management.